Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO  uBio
uBio Compartir

 Permalink
PermalinkRevista Boliviana de Química
versión On-line ISSN 0250-5460
Rev. Bol. Quim vol.33 no.2 La Paz jul. 2016
REVIEWS AND THEORETICAL ARTICLES
Synthesis of alkenes: Claisen rearrangement of allyl vinyl ethers, part II; Mechanistic views; The organic chemistry notebook series, a didactical approach, N° 10
Síntesis de alquenos mediante transposición de Claisen de éteres alil-vinílicos, parte II; vistas mecanicísticas; de la serie: El cuaderno de notas de química orgánica, un enfoque didáctico, N°10
José A. Bravo1,*, José L. Vila2
1 Department of Chemistry, Laboratorio de Fitoquímica, Instituto de Investigaciones en Productos Naturales IIPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, Phone 59122792238, La Paz, Bolivia, jabravo@umsa.bo
2Department of Chemistry, Laboratorio de Síntesis y Hemisíntesis, Instituto de Investigaciones en Productos Naturales IIPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, Phone 59122795878, La Paz, Bolivia, joselu62@hotmail.com
*Corresponding author: joseabravo@outlook.com
Abstract
This is the tenth theoretical assay in the series: "The Organic Chemistry Notebook Series, a Didactical Approach".
The aim of this series of studies is to help students to have a graphical view of organic synthesis reactions of diverse nature. We have taken a series of reactions compiled by W. Carruthers in 'Some modern methods of organic synthesis', and we have proposed didactical and mechanistic views for them. This theme is included in the chapter "Formation of carbon-carbon double bonds" in the mentioned text.
In the present chapter, we expose a complementing of Claisen rearrangements of ally-vinyl ethers. A first approach in this study comports the Claisen rearrangement feature regarding the stereochemical control that conducts to the definition of the positioning of substituents on the new single bond that arises from rearrangement. This feature serves as a sort of transmission of chirality along a carbon chain. We proposed mechanisms for this feature. As an example we elaborated mechanisms for a key step in the synthesis of (+)-15(S)-prostaglandin A2 involving the stereochemical control mentioned. We also covered from a mechanistic stand point the theme of the geometry of the enol ether double bond and its control by means of the procedure described by Ireland. We elaborated too, the mechanisms for the conversion of allylic ester into the E-ketene acetal or the Z-ketene acetal and subsequent acid formation including the example of the E-crotyl propanoate and the Z-crotyl propanoate.
Keywords: Organic Chemistry, Alkenes, Allyl vinyl ethers, Claisen rearrangement, Mechanisms of Reactions, W. Carruthers.
Resumen
Este es el décimo ensayo teórico en la serie: "El cuaderno de química orgánica, un enfoque didáctico".
El objetivo de esta serie de estudios es ayudar a los estudiantes a disponer de una visión gráfica de reacciones de síntesis orgánicas de diversa naturaleza. Hemos tomado una serie de reacciones compiladas por W. Carruthers en: 'Some modern methods of organic synthesis', para las cuales hemos propuesto vistas mecanicísticas y didácticas. Este tema esta incluido en el capítulo "Formation of carbon-carbon double bonds" del mencionado texto.
En el actual capítulo, abordamos un complemento de la transposición de Claisen de éteres alil-vinílicos. Un primer enfoque en este estudio comporta el sobresaliente hecho de la transposición de Claisen que guarda relación con el control estereoquímico que conduce a la definición de la posición de los sustituyentes en el nuevo enlace simple que se forma de la transposición. Este hecho remarcable sirve como una especie de transmisión de quiralidad a lo largo de la cadena carbonada. Hemos propuesto mecanismos para este hecho. Como un ejemplo hemos elaborado mecanismos para el paso clave en la síntesis de (+)-15(S)-prostaglandin A2 que involucra el mencionado control estereoquímico. También, hemos cubierto mecanicísticamente el tema de la geometría del doble enlace del
enol eter y su control por medio del procedimiento descrito por Ireland. También hemos elaborado los mecanismos para la conversión de éster alílico en E-ceteno acetal o en Z-ceteno acetal y su subsecuente conversión en ácido incluyendo el ejemplo del propanoato de E-crotyl y de Z-crotyl.
INTRODUCTION
During master classes of organic chemistry we noticed that students are confronted with a lack of knowledge with regard to mechanisms. For instance, oxidation-reduction reactions which are among the most commonly employed constitute a kind of black box for the student's mind. A mechanistic approach about any kind of reaction enhances the capacity of facing new reactions with respect to an understanding of all processes involved in them, and also develops synthetic creativity. As academics we feel concerned with the didactical importance of covering these needs in debutant students in organic synthesis. This, the synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part II; mechanistic views; is the tenth study in the series: "The Organic Chemistry Notebook Series, a Didactical Approach" [1-9].
REACTIONS AND THEIR MECHANISTIC PROPOSALS, DISCUSSION
The Claisen rearrangement of allyl vinyl ethers constitutes a stereoselective way to the production of j^unsaturated aldehydes, as well as ketones, esters and amides [9,10]. The Claisen rearrangement comports a feature regarding the stereochemical control that conducts to the definition of the positioning of substituents on the new single bond that arises from rearrangement [11]. At least three chiral centers may be involved in the rearrangement. One in the starting reagent 1 or 3, and at least one in the product 2 [11]. The reaction takes place in a suprafacial manner with respect to the allylic part [11]. The configuration at C-3 of the product (2 or 4) comes from directly from the starting reagent [11], see Fig. 1. Any of the configurations at the chiral carbon formed (C-3 in 2 or 4) can be obtained if a change in the stereochemistry of double bond of the allylic portion is done. The oxygenated chiral carbon in 1 loses chirality in 2 and 4 after the reaction takes place [11]. A new chiral centre appears in the product at the allylic position in a controlled manner [11]. This feature serves as a sort of transmission of chirality along a carbon chain [11].

Comments
The driving force in all pericyclic rearrangement including the Claisen rearrangement results from a contest between the ring strain in the transition state that should lower and the tendency to form the carbonyl double bond that should increase. The formation of the carbonyl double bond is favored by the fact that an enol or an enolate is less stable than a carbonyl even if it is not a a/?-unsaturated but a j#unsaturated carbonyl compound.
Let us survey the key step in the synthesis of (+)-15(S)-prostaglandin A2 for example [11]. In this step, the allylic alcohol 5, optically active, is transformed into the j#unsaturated ester 6, optically active following the orthoester procedure [11,12]. See Fig. 2.

Comments
Figure 2 shows the nucleophilic attack starred by the oxygen of the allylic alcohol 5 over the trimethoxyalkyl as substrate. The formed adduct undergoes an elimination of methanol to form the enolate. This adducts suffers the Claisen rearrangement to afford the j#unsaturated ester 6.
There is a correlation between chiral centers at C-2 and C-3 in compounds 2 and 4 (Fig. 1) which is derived from the chair type transition state and depends on the geometry of the double bonds in the starting material [11]. Fig. 3 shows this concept for the isomeric 2-butenyl-1-propenyl ethers (7 and 10) [11]. By choosing one or another geometry in the double bonds of the adduct enol + alkene, namely the vinyl ether, it is feasible after the Claisen rearrangement, the obtaining of the two configurations for one of the chiral centers, keeping unaltered the configuration of the vicinal chiral center. This is the case for C*-2 in 2-butenyl-1-propenyl ether (Fig. 3) and for C*-3 in Fig. 1 [11]. Also let us remark the dependence on the temperature with regard to the stereochemistry during the Claisen rearrangement, as shown in Fig. 3.
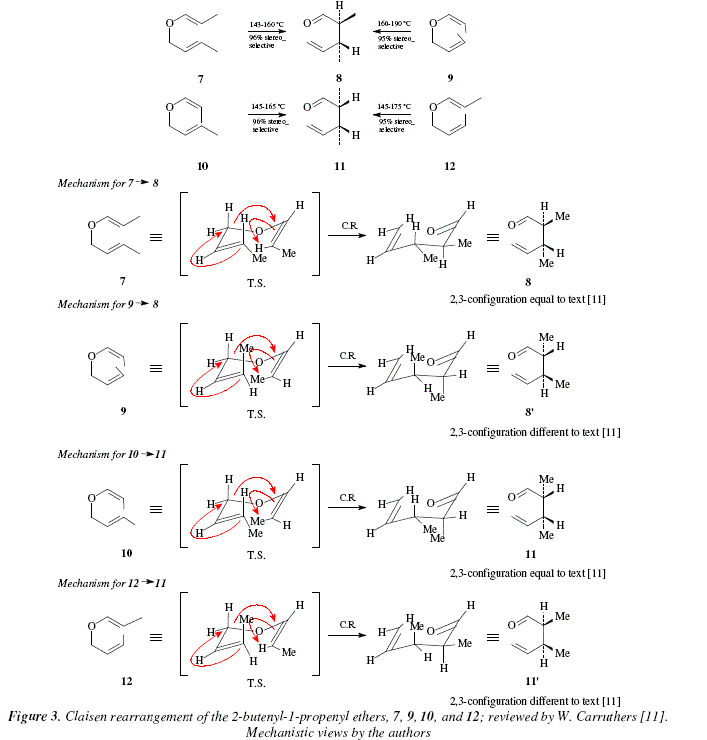
Comments
In Fig. 3, only the products 8 or 11 were expected by either method, by using four different starting molecules, namely, 2-butenyl-1-propenyl ethers, exhibiting different stereochemistry in the double bonds (7, 9, 10, and 12) at low and higher temperature. However, according to the suprafacial rearrangement with a chair-type transition state at 160-190° (when employing 9) and 145-175° (when employing 12), the product was not the expected as when using 7 and 10 with a transition state of the chair-type . The reason to obtain 8 and 11 instead of 8' and 11' could be that the higher temperature makes possible an isomerization ZE in 9 and 12 in order to comply with the stereochemistry defined by the transition state in the same manner as when the starting molecules are 7 and 10.
The Ireland's ester-enolate procedure described previously [9,11] is considered among the best methods when controlling the geometry of the double bond of the enol ether [11,13]. In this sense, the conversion of the allylic ester 13 into the silyl ketene acetal by using lithium diisopropylamine in THF with subsequent silylation with chlorodimethylterbutyl silane, usually conducts to the E-ketene acetal 14 and later to its acid 15 [11]. See Fig. 4.

Conversely, when lithium diisopropylamine in THF is used mixed with 23% of hexamethyl-phosphoric triamide (a polar co-ordinating solvent) and subsequent silylation, the reaction conducts to the Z-ketene acetal 16 and the acid 17 [11].See Fig. 5.

Comments
In Fig. 4 and 5, the strong base LiN(iso-C3H7)2 is the responsible for the removal of the a proton of carbonyl. The electronic excess over the a carbon is stabilized with the cation Li+. Is at this stage that the stereochemistry EorZof the double bond of the enol moiety of the enol ether will be defined depending on the solvent (lithium diisopropylamine in THF or lithium diisopropylamine in THF plus hexamethyl-phosphoric triamide respectively according to Ireland). There is after, a transferring of charge from the carbanion-lithium to the oxygen of the carbonyl to form a new oxygen-lithium pair. The Lewis acid Li+ is weaker than the terbutyldimethylsilyl cation which gives a better stability to the adduct to evolve and adopt the chair-type transition state propitious for the Claisen rearrangement. The rearrangement affords the j^unsaturated silyl ester first and the ulterior γδunsaturated carboxylic acid, after catalytic protic hydrolysis.
For example, let us survey the E-crotyl propanoate 18, which produces principally the erythro acid 19 when the enolisation is done in THF, however, the threo form 20 predominates if the solvent THF contains hexamethyl-phosphoric triamide [11,14]. When the Z-crotyl propanoate 21 [14] is employed, the opposite stereochemistry is observed [11,14]. See Fig. 6 and 7.



In the case of using THF without HMPA at -78°C (not so co-ordinating solvent), the ester carbonyl group in association with lithium is important and the carbonyl exhibits a stronger steric effect than the oxygen from ether [11]. Non-bonding interactions favor a transition state like 22 (a) and consequently the formation of the Z-enolate is favored. If HMPA is added to the solvent, co-ordination is less important and the ether oxygen is the more demanding sterically, consequently, the E-enolate is favored [11]. See Fig. 8 and 9.


ACKNOWLEDGEMENT
The authors express their gratitude to Prof. Eduardo Palenque from the Department of Physics, Universidad Mayor de San Andrés, for his bibliographic support.
REFERENCES
1. Bravo, J. 2005, The organic chemistry notebook series, a didactical approach. Theoretical mechanistic approach to diasteroselective synthesis of cis-1,2-dialkenylcyclopropanols and subsequent oxy-Cope rearrangement by Jin Kun Cha et al, Rev. Bol. Quim., 23, 1-10. [ Links ]
2. Bravo, J.A., Mollinedo, P., Peñarrieta, J.M., Vila, J.L. 2013, Mechanistic views of intramolecular hydroxycyclopropanation of ¿y-vinyl carboxylic esters, Rev. Bol. Quim., 30 (1), 24-41. [ Links ]
3. Bravo, J.A., Vila, J.L. 2014, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part I; the organic chemistry notebook series, a didactical approach, n° 3. Rev. Bol. Quim., 31 (1), 61-67. [ Links ]
4. Bravo, J.A., Vila, J.L. 2015, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part II; the organic chemistry notebook series, a didactical approach, n° 4, Rev. Bol. Quim., 32 (1), 15-23. [ Links ]
5. Vila, J.L., Bravo, J.A. 2015, Synthesis of alkenes by fragmentation reactions; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 5, Rev. Bol. Quim., 32 (2), 37-44.
6. Bravo, J.A., Vila, J.L. 2015, Synthesis of alkenes by oxidative decarboxylation of carboxylic acids; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 6, Rev. Bol. Quim., 32 (3), 45-52. [ Links ]
7. Bravo, J.A., Vila, J.L. 2015, Synthesis of alkenes from ketones via arylsulphonyl-hydrazones; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 7, Rev. Bol. Quim., 32 (4), 82-89. [ Links ]
8. Bravo, J.A., Vila, J.L. 2015, Stereospecific synthesis of alkenes from 1,2-diols; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 8, Rev. Bol. Quim., 32 (5), 121-125. [ Links ]
9. Bravo, J.A., Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part I; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 9, Rev. Bol. Quim., 33 (1), 27-33. [ Links ]
10. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 167-169. [ Links ]
11. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 170-172. [ Links ]
12. Stork, G, Raucher, S. 1976. Chiral synthesis of prostaglandins from carbohydrates. Synthesis of (+)-15-(S)-prostaglandin A2. J. Am. Chem. Soc. 98, 1583-1584. [ Links ]
13. Ireland, R.E., Mueller, R.H., Willard, A.K. 1976, The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation, J. Am. Chem. Soc., 98 (10), 2868-2877. [ Links ]
14. Bohlmann, R., Brown J.M., Brunner H., Davis A.P., El-Khawaga M. et al. D.1.6. Formation of C-CBonds by Pericyclic Reactions In: Houben-Weyl Methods of Organic Chemistry Vol. E 21d, 4th Edition Supplement: Stereoselective Synthesis: C-C Bond Formation by Sigmatropic Rearrangements, Electrocyclic Reactions, C-H, C-Hal Bond Formation, ed by Helmchen, G., Hoffmann, R.W., Mulzer, J., Schaumann, E. 2014, Georg Thieme Verlag, Stuttgart, pp. 3412-3414. [ Links ]