Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO  uBio
uBio Compartir

 Permalink
PermalinkRevista Boliviana de Química
versión On-line ISSN 0250-5460
Rev. Bol. Quim vol.32 no.4 La Paz nov. 2015
ARTÍCULOS ORIGINALES
Synthesis of alkenes from ketones via arylsulphonyl-hydrazones; mechanistic views; the organic chemistry notebook series, a didactical approach, n27
José A. Bravo1*, José L. Vila2
1 Department of Chemistry, Laboratorio de Fitoquímica, Instituto de Investigaciones en Productos Naturales IIPN,
Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota,
Phone 59122792238, La Paz, Bolivia, jabravo@umsa.bo
*Corresponding author: joseabravo@outlook.com
2Department of Chemistry, Laboratorio de Síntesis y Hemisíntesis, Instituto de Investigaciones en Productos Naturales IIPN,
Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota,
Phone 59122795878, La Paz, Bolivia, joselu62@hotmail.com
Abstract
This is the seventh chapter in the series published by the same authors: "The Organic Chemistry Notebook Series, a Didactical Approach".
The aim of this series of studies is to help students to have a graphical view of organic synthesis reactions of diverse nature. Here we describe the mechanistic views of the synthesis of alkenes from ketones via arylsulphonylhydrazones. These methods employ aliphatic and alicyclic ketones with one a-hydrogen, that react along with toluene-p-sulphonylhydrazones and two equivalents of an alkyl-lithium or lithium diisopropylamide. The mechanism views for the transformation of pinacolone into 3,3-dimethyl-1-butene are proposed. The formation of 3-phenylpropene using phenylacetone is explained step by step. An approach is made on the obtaining of alkenes from ketones using derived enol ethers or esters by means of reductive excision or by means of coupling with organocuprates. We have used various series of reactions reviewed by W. Carruthers in 'Some modern methods of organic synthesis', and we have proposed didactical and mechanistic views for them. This theme is included in the chapter "Formation of carbon-carbon double bonds" in the text mentioned above. Spanish title: Síntesis de alquenos a partir de cetonas via arilsulfonilhidrazonas; vistas mecanísticas; De la serie: El cuaderno de notas de química orgánica, un enfoque didáctico, N°7.
Keywords: Organic Chemistry, Ketones, Arylsulphonylhydrazones, Alkenes, Mechanisms of Reactions, W. Carruthers.
INTRODUCTION
During master classes of organic chemistry it is possible to notice that students are confronted with a lack of knowledge with regard to mechanisms. For instance, oxidation-reduction reactions which are among the most commonly employed constitute a kind of black box for the student' s mind. A mechanistic approach of any kind of reaction enhances the capacity of facing new reactions with respect to an understanding of all processes involved in them, and also develops synthetic creativity. As academics we feel concerned with the didactical importance of covering these needs in debutant students in organic synthesis. This, the synthesis of alkenes from ketones via arylsulphonylhydrazones, is the seventh study in: "The Organic Chemistry Notebook Series, a Didactical Approach" [1-6].
REACTIONS AND THEIR MECHANISTIC PROPOSALS, DISCUSSION
Aliphatic and alicyclic ketones with at least one hydrogen in ![]() are employed to synthesize alkenes [7]. The method includes reaction of ketones with toluene-p-sulphonylhydrazones and the addition of two equiv of lithium diisopropylamide or alkyl lithium [7-9]. Products present no changes in the carbon framework, and given the adequate mild conditions; the less substituted alkene is obtained [7]. Pinacolone gives an only product: 3,3-dimethyl-1-butene [7]. See Fig. 1 and 2, and Fig. 3 for a mechanistic explanation. 3-phenylpropene is brought forth by phenylacetone, however no styrene is present after reaction [7] (Fig. 1, 2 and Fig. 4).
are employed to synthesize alkenes [7]. The method includes reaction of ketones with toluene-p-sulphonylhydrazones and the addition of two equiv of lithium diisopropylamide or alkyl lithium [7-9]. Products present no changes in the carbon framework, and given the adequate mild conditions; the less substituted alkene is obtained [7]. Pinacolone gives an only product: 3,3-dimethyl-1-butene [7]. See Fig. 1 and 2, and Fig. 3 for a mechanistic explanation. 3-phenylpropene is brought forth by phenylacetone, however no styrene is present after reaction [7] (Fig. 1, 2 and Fig. 4).

Various steps are applicable to the reaction. First, the anion 1 is formed by action of the first alkyl lithium [7]. Then the second equivalent of alkyl lithium generates another charge and the dianion 2 appears. Localization of the excessive charge in the carbanion of 2, leads to the fragmentation of the arylsulphonyl moiety [7]. The resulting anionic diazo-alkene becomes a carbanion by means of excision of one molecule of gaseous nitrogen; the carbanion makes a new covalence with one cation Lithium, the other one compensates the charge of the arylsulphonate [7]. The yield can be improved by using tetramethylethylenediamine (TMEDA) [7] as solvent. TMEDA is the additive or solvent necessary to obtain the less substituted alkene from an unsymmetrical ketone [7]. When an ethereal or hydrocarbon solvent is used, the C=C bond occupies a position defined by the stereochemistry of the hydrazone [7]. The second abstraction of the a proton in 1, happens in a syn way to the ![]() [7,10].
[7,10].
Intermediate 3 can suffer interchange of the cation Li+ with H+ of the solvent (or an alternative source of protons) or being substituted by another electrophile [7]. See Fig. 2 and Fig. 3.

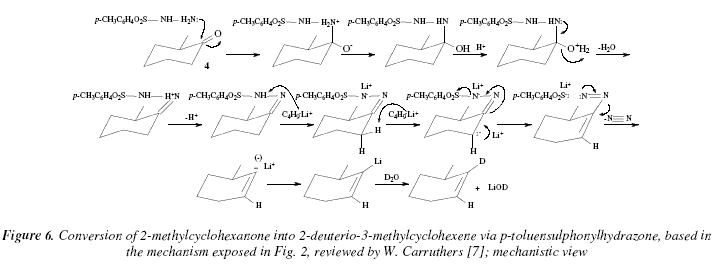
In TMEDA, submitting the reaction to the adding of deuterium oxide is a way to deuterated alkenes (Fig. 1 and Fig. 4) [7]. 2-methylcyclohexanone (4, Fig. 5 and 6), gives rise to 2-deuterio-3-methyl cyclohexene, and octan-2-one (5, Fig. 5 and 7) to 2-deuterio-1-octene [7]. The preferred form of the 1,2-disubstituted alkene after protonation of the lithium intermediate 3 (Fig. 2) is the Z-isomer [7].
The intermediates like 3 (Fig. 2), can be usually treated with other electrophiles like dibromoethane for instance, to give rise to vinyl bromides, or acetaldehyde to give allyl alcohols or carbon dioxide to give acrylic acids [7] (Fig. 5 and 7). The hydrazone more adequate in these reactions is the 2,4,6-triisopropylbenzenesulphonylhydrazone to provide the vinyl-lithium, instead of the p-toluensulphonyl [7]. This latter hydrazone suffers lithiation at the ortho position of the aromatic ring, provoking thus a diminution of the yields in the substituted alkene [7]. This fact is avoided using the triisopropyl derivative [7,11,12]; see Fig. 5 and 7.



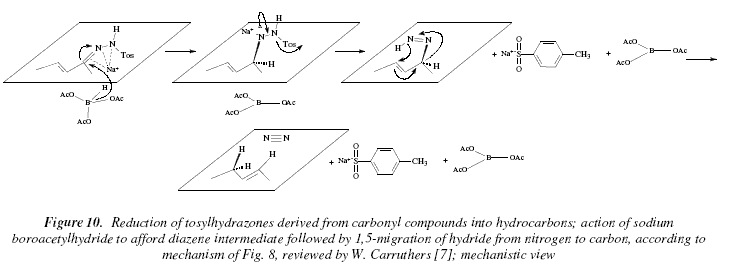
This can be achieved by using sodium cyanoborohydride in acid [7]. An alternative to the Wolf-Kishner deoxygenation is the using of catecholborane [7]. When the hydrazones derivatives are ![]() unsaturated, it is possible to form alkenes where the double bond has moved to the original carbonyl position [7]. The reducing agent is usually NaBH(OCOCH3) coming from the mixture: NaBH4 and acetic acid [7,13]. A tentative mechanism has been proposed where the formation of the diazene intermediate suffers a migration of hydride (15) from N to C [7]. See Fig. 8, 9 and 10.
unsaturated, it is possible to form alkenes where the double bond has moved to the original carbonyl position [7]. The reducing agent is usually NaBH(OCOCH3) coming from the mixture: NaBH4 and acetic acid [7,13]. A tentative mechanism has been proposed where the formation of the diazene intermediate suffers a migration of hydride (15) from N to C [7]. See Fig. 8, 9 and 10.



Moreover, the use of sodium borodeuteride/acetic acid [NaBD4 + 3CH3COOH^ NaBD(CH3COO-)3], or on the other hand the use of carboxyl deuterated acetic acid/sodium borohydride, conducts to the regio-selective introduction of deuterium (one or two units) [7]. When deuterated acetic acid is employed, the interchange at the N-H position with deuterium should be faster than the process of reduction (hydride transfer toward carbon) [7]. See Fig. 11, 12 and 13.



Ketones are also precursors of alkenes by the way of derived enol ethers or enol esters [7]. This is achieved by reductive cleavage or by coupling with organocuprates [7]. The substrate to be reduced by the using of lithium and amines is the N,N,N',N'-tetramethylphosphordiamidates. The cleavage occurs on the C-O bond [7]; with enol derivatives it conducts to the corresponding alkene with good yields [7,14]. See Fig. 14 and 16.

The enol can be reduced, it means see its oxygen replaced by an anion, rather by a carbanion (alkyl), instead of a hydride (a basic hydrogen), by the reaction of the enol diphenylphosphate or the enol trifluoromethanesulphonate (triflate) with lithium dialkylcuprate [7,15,16]. See Fig. 15. The high stereoselectivity characterizes these reactions [7]. As an example let us examine the transformation of (Z)-5-trifluoromethanesulphonyloxy-5-decene into (E)-5-methyl-5-decene by using dimethyl cuprate [7]. See Fig. 15


In Fig. 16 the basic nitrogen temporally stabilized by the cation Li+, withdraws the proton in ![]() of carbonyl in the ketone. This is due to its acidic character. A carbanion appears in the place of the a-hydrogen. Because the oxygen supports better than the carbon atom a negative charge in excess, an inductive current generates the migration of the two electrons over the carbon atom for establishing a new C2=C3 double bond and placing the negative charge over the oxygen of the ketone now transformed into an enol oxygen. Oxygen is now temporally stabilized by Li+. This lithium enol interacts now with chloride from phosphorus derivative {[(CH3)2N]2POCl} thus generating the corresponding cation with the positive charge reposing on the phosphorus. What is next is the oxidation of two equiv. of Lithium metallic plus the using of two equiv. of ammonia as well as two equiv. of n-butanol to give one ammonia, a n-butoxylammonium, a lithium-n-butoxyl and a lithium hydride. This hydride is responsible of the reduction of the enol into an alkene function.
of carbonyl in the ketone. This is due to its acidic character. A carbanion appears in the place of the a-hydrogen. Because the oxygen supports better than the carbon atom a negative charge in excess, an inductive current generates the migration of the two electrons over the carbon atom for establishing a new C2=C3 double bond and placing the negative charge over the oxygen of the ketone now transformed into an enol oxygen. Oxygen is now temporally stabilized by Li+. This lithium enol interacts now with chloride from phosphorus derivative {[(CH3)2N]2POCl} thus generating the corresponding cation with the positive charge reposing on the phosphorus. What is next is the oxidation of two equiv. of Lithium metallic plus the using of two equiv. of ammonia as well as two equiv. of n-butanol to give one ammonia, a n-butoxylammonium, a lithium-n-butoxyl and a lithium hydride. This hydride is responsible of the reduction of the enol into an alkene function.
ACKNOWLEDGEMENTS
The authors express their gratitude to Prof. Eduardo Palenque from the Department of Physics, Universidad Mayor de San Andrés, for his bibliographic support.
REFERENCES
1. Bravo, J. 2005, Rev. Bol. Quim., 23, 1. [ Links ]
2. Bravo, J.A., Mollinedo, P., Peñarrieta, J.M., Vila, J.L. 2013, Rev. Bol. Quim., 30, 24. [ Links ]
3. Bravo, J.A., Vila, J.L. 2014, Rev. Bol. Quim., 31, 61. [ Links ]
4. Bravo, J.A., Vila, J.L. 2015, Rev. Bol. Quim., 32, 15. [ Links ]
5. Vila, J.L., Bravo, J.A. 2015, Rev. Bol. Quim., 32, 37. [ Links ]
6. Bravo, J.A., Vila, J.L. 2015, Rev. Bol. Quim., 32, 45. [ Links ]
7. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 162-165. [ Links ]
8. Kolonko K.J., Shapiro, R.H. 1978, J. Org. Chem., 43, 1404. [ Links ]
9. Adlington, R.M., Barrett, A.G.M. 1983, Acc. Chem. Res., 16, 55. [ Links ]
10. Lipton, M.F., Shapiro, R.H. 1978, J. Org. Chem., 43, 1409. [ Links ]
11. Chamberlin, A.R., Stemke, J.E., Bond, F.T. 1978, J. Org. Chem., 43, 147. [ Links ]
12. Chamberlin, A.R., Liotta, E.L., Bond, F.T. 1983, Organic Syntheses, 61, 141. [ Links ]
13. Hutchins, R.O., Natale, N.R. 1978, J. Org. Chem. 43, 2299. [ Links ]
14. Ireland, R.E., O'Neil, T.H., Tolman, G.L. 1983, Organic Syntheses, 61,116. [ Links ]
15. McMurry, J.E., Scott, W.J. 1980, Tetrahedron Lett. 21, 4313. [ Links ]
16. McMurry, J.E., Scott, W.J. 1980, Tetrahedron Lett. 24, 979. [ Links ]