Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkRevista Boliviana de Química
versión On-line ISSN 0250-5460
Rev. Bol. Quim vol.34 no.2 La Paz 2017
ARTÍCULOS DE REVISIÓN
Claisen rearrangement of allyl vinyl ethers to afford alkenes,
Part IV; mechanistic theoretical proposals; the organic chemistry notebook series, a
Didactical approach, N° 12
Reordenamiento de claisen de eteres alil vinilicos para dar alquenos,
Parte IV; propuestas mecanicistas teoricas; serie el cuaderno de quimica organica,
Un enfoque didactico, N° 12
Jose A. Bravo1,*, Jose L. Vila2
1 Natural Product Laboratory, Phytochemistry, Chemical Sciences Dep., School of Pure and Natural Sciences FCPN,
Universidad Mayor de San Andres UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota,
phone +59122792238, La Paz, Bolivia, jabravo@umsa.bo, www.umsa.bo
2Natural Product Laboratory, Hemisynthesis and Green Chemistry, Chemical Sciences Dep.,
School of Pure and Natural Sciences FCPN, Universidad Mayor de San Andres UMSA, P.O. Box 303, Calle Andres Bello s/n,
Ciudad Universitaria Cota Cota, phone +59122772269, La Paz, Bolivia, jlvila@umsa.bo, www.umsa.bo
*Corresponding author: jabravo@umsa.bo
Received 10 19 2016 Accepted 06 29 2017 Published 06 30 2017
ABSTRACT
This is the twelfth theoretical assay in the series: "The Organic Chemistry Notebook Series, a Didactical Approach".
The aim of this series of studies is to help students to have a graphical view of organic synthesis reactions of diverse nature. We have taken a series of reactions compiled by W. Carruthers in 'Some modern methods of organic synthesis', and we have proposed didactical and mechanistic views for them. This theme is included in the chapter "Formation of carbon-carbon double bonds" in the mentioned text.
In the present chapter we expose more Claisen rearrangements variations. The Claisen rearrangement can be regarded as a particular case of Cope's rearrangement, hence, on the basis of the structural theory and the known mechanisms of organic chemistry, we have proposed theoretical mechanisms for the synthesis of dienes from 2-methyl-3-phenyl-1,5-hexadiene at low temperature. We have proposed a mechanism for the synthesis of δe-unsaturated aldehydes and ketones through the oxy-Cope rearrangement of 1,5-hexadienes. We've described the mechanism for the key step of the synthesis of the sesquiterpene (±)-juvabione. The mechanistic views of the synthesis of the natural germacrane sesquiterpenes are proposed. We have proposed a mechanism for the thio Claisen rearrangement of allyl vinyl sulfides in the transformation into ƴδ5-unsaturated carbonyl compounds, for example the preparation of 4-tridicenal and the [2,3]sigmatropic rearrangement of allylsulphonium ylids like in the preparation of y-cyclocitral.
Keywords: Organic Chemistry, Alkenes, Allyl vinyl ethers, Claisen rearrangement, Mechanisms of Reactions, W. Carruthers.
RESUMEN
Spanish title: Reordenamiento de Claisen de éteres alíl vinílicos para dar alquenos: parte IV; propuestas mecanicistas teóricas; serie el cuaderno de química orgánica, un enfoque didáctico, N°12. Este es el duodécimo ensayo teórico en la serie: "El cuaderno de química orgánica, un enfoque didáctico".
El objetivo de esta serie de estudios es ayudar a los estudiantes a disponer de una visión gráfica de reacciones de síntesis orgánicas de diversa naturaleza. Hemos tomado una serie de reacciones compiladas por W. Carruthers en: 'Some modern methods of organic synthesis', para las cuales hemos propuesto vistas mecanicistas y didácticas. Este tema está incluido en el capítulo "Formation of carbón-carbón double bonds" del mencionado texto.
En el presente capítulo exponemos más variaciones del reordenamiento de Claisen. El reordenamiento de Claisen puede ser considerado como un caso particular del reordenamiento de Cope, por lo tanto, hemos propuesto mecanismos teóricos basados en la teoría estructural y los mecanismos conocidos de la química orgánica, para la síntesis de dienos a partir de 2-metil-3-fenil-1,5-hexadieno a bajas temperaturas. Hemos abordado por mecanismos la síntesis de aldehidos y cetonas insaturados en δe a través de transposiciones oxy-Cope de 1,5-hexadienos. Hemos descrito el mecanismo del paso clave de la síntesis del sesquiterpeno (±)-juvabione. Las vistas mecanicistas de la síntesis de los productos naturales sesquiterpenos del germacrane se hallan propuestas. Hemos propuesto un mecanismo para la transposición del tipo tio-Claisen de sulfuras de alilivinilo en la transformación en compuestos carbonilo ƴδ-insaturados, como la preparación del 4-tridecenal y el reordenamiento [2,3]-sigmatrópico de ilidos alilsulfonios como en la preparación de y-ciclocitral.
INTRODUCTION
Master classes of organic chemistry showed us the difficulties experimented by students due to lack of knowledge of classical mechanisms. A mechanistic proposal is naturally mandatory if a rational explanation of products emerging from a synthesis is going to be formally accepted and understood. As academics we are committed with the didactics and we have designed a series of articles exposing mechanistic theoretical proposals, articles have a character of review, meaning thus the use of published works on varied themes on synthesis. The present contribution: mechanistic theoretical proposal of Claisen rearrangement of allyl vinyl ethers to afford alkenes, part IV, is the twelfth study in the series: "The Organic Chemistry Notebook Series, a Didactical Approach" [1-11].
REVIEW AND DISCUSSION BY MEANS OF MECHANISTIC THEORETICAL PROPOSALS
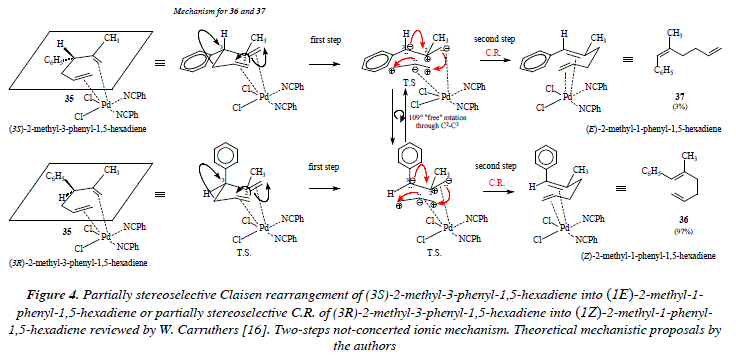
The Claisen rearrangement of allyl vinyl ethers can be considered as one case of the Cope [3,3] -sigmatropic rearrangement of 1,5-hexadienes [1,12,13,14]. See Figure 1. In laboratory, the Cope rearrangement requires high temperatures to proceed [12]. More recently, experiments demonstrated that the temperature factor can be dramatically diminished (until room temperature) by means of the use of catalytic amounts of palladium chloride bis(benzonitrile) complex [12,15]. As an example let's mention the use of 2-methyl-3-phenyl-1,5-hexadiene 35 with a catalytic quantity of PdCl2(PhCN)2 (in THF, rt, 24 hs.) to synthesize the dienes 36 and 37; yield was 87% and the ratio was: 97:3. The change of THF for benzene conducted the reaction in one hour instead of 24 hs. On the contrary the Cope rearrangement (under thermal conditions) of 35 gave rise to products poor in stereoselectivity and with the inconvenience of the elevate temperature [16]. See Figures 2 and 3 for the reaction scheme and the corresponding mechanistic proposals.

Comments
Figure 2 shows the hypothetical fully stereoselective synthesis of (lE)-2-methyl-l-phenyl-1,5-hexadiene (37) and (lZ)-2-methyl-l-phenyl-1,5-hexadiene (36) from (3S) and (3R)-2-methyl-3-phenyl-1,5-hexadiene (35), respectively. This scheme (Fig. 2) is only for didactical purposes, to show that 37 comes from (S)-35 and 36 from (R)-35, hypothetically considering that the Claisen rearrangement is completely stereoselective en each case. However, the departure from any of the enantiomeric forms of 35 gives both stereochemical isomers: 36 and 37 in different proportions. This fact implies the fact of free rotation between any of the forms conducting to the positioning of the phenyl group in an axial or in an equatorial orientation. On the one hand, this means that the transition state in the chair form of the C.R. clearly exhibits the partial existence of the new bonds being formed (o and π) and the partial disintegration of old bonds (o and π). In such status, when the overlapping of p orbitals of the forming or the dis-overlapping of the disappearing π bonds is not completed, the rotation around a sigma bond is feasible. This situation makes possible the rotation of the phenyl substituent at C-3. The rotation is feasible around the C2-C3 bond, once partially disappeared the bond C3-C4 and no completely formed the π bond between the C2-C3 bond of 35. The transition state of the partially stereoselective C.R. of 35 is described in Fig. 3. We must notice (Fig. 3) that the fact of being in the presence of a pericyclic reaction, implies, by the way, that in order to impulse the rearrangement there must be the appropriate degree of overlapping between p contiguous orbitals.


This fact is indeed, an impeachment to free rotation around bonds. On the other hand, if the character of the rearrangement is not pericyclic, it's then ionic. Under the scope of the formation of ions (anion and cation) in the intermediates of the ionic transition state, rotation is easier than in the concerted pericyclic mechanism (there is not overlapping of contiguous p orbitals). If instead of a concerted one-step rearrangement, we propose an ionic one, then we have a not-concerted two-steps process. Let us notice as well that free rotation in the ionic transition state does not forcedly implies a full stereoselectivity and that we must contemplate the possibility of a not necessarily equal percentage distribution of the stereo isomers: (lE)-2-methyl-l-phenyl-l,5-hexadiene (37) and (lZ)-2-methyl-l-phenyl-l,5-hexadiene (36) (a racemic mixture). This theoretical approach seems to suggest that the Claisen rearrangement of (3S)-2-methyl-3-phenyl-l,5-hexadiene or of (3R)-2-methyl-3-phenyl-l,5-hexadiene does not proceed via a concerted one-step pericyclic transition state but instead via a two-steps not-concerted ionic transition state. See Fig. 4. The ionic transition state in Fig. 4 is firstly stabilized by the catalyst palladium and secondly by mutual electrostatic attraction between opposed charges in both, the propene, and the 2-methyl-3-phenyl-propene moieties. From experimental results it is evident that the speed of the electronic movement in the hypothetical ionic transition state that conducts to the Claisen compound is higher to that which conducts to the formation of the two much tensioned fused cicyclobutanes derived from the coupling of opposite charges.
The rearrangement of 3-hydroxylated 1,5 dienes known as the oxy-Cope rearrangement [1] is suitable for the synthesis of <5e-unsaturaded ketones or aldehydes (See Fig. 5). When positions 3 and 4 of the diene are hydroxylated, the product is a 1,6-dicarbonyl compound. See Fig. 5. The rearrangements can be made much faster by the adding of KOR instead of KOH; the metal ion (K+), once ionized and solvated and free of the oxyanion [17,18,19]. Rearrangements follow a chair-like (or a boat-like) transition state giving high stereoselectivity [17]. The diene 38 transforms into 39, an unsaturated ketone. The isomer 40 was not touched. See Fig. 6.


Comments
The driving force in the oxy-Cope rearrangement of 38 into 39 is due to the stereochemical form in 38 (Fig. 6) with the terminal vinyl with an α stereochemistry (axial position) inducing the electronic attack coming from the cyclic alkene. Such terminal vinyl axial position is propitious for the opposite charges attraction that appears once the boat-like transition state is formed. Thus the one-bond distance in the space between opposite charges makes possible the expected or observed cyclisation, or compound 39. Let us signal at this point that the isomer 40 would exhibit a carbanion extreme (developed over the terminal vinyl), placed too far away from the carbocation making thus impossible the opposite charges attraction in the bipolar species by means of the covalence formation. This is the reason for the isomer 40 to rest untouched in the oxy-Cope rearrangement.
The synthesis of (racemic)-juvabione, a natural sesquiterpene, biomimetic of the juvenile hormone [20], shows its key step where the diene 41 affords the cyclohexanone with high stereoselectivity (77% yield) [17,19]. See Fig. 7.

Comments
The side chain of the cyclohexene 41 accommodates in such a manner with respect to the cycle that together form a boat-like transition state, with the double bonds face-to-face. This is the only way to provoke a rearrangement; the mechanism in Fig. 7 shows two forms of the transition state in which negative and positive charges are localized. These two forms present the terminal methyl group in opposite sides. One converts into another by free rotation around the bond between the carbocation and its vicinal sp2 carbon. However, the real mechanism is supposed to be pericyclic. Anyway, the free rotation is feasible only under the perspective of localized charges instead of overlapping orbitals p. Here again, we find two contradicting concepts inherent to rearrangements as previously exposed in this paper (cf. Fig. 3 and 4).
This mechanism is applicable to any other similar synthesis example, like the synthesis of sesquiterpenes v.g. the synthesis of germacrenes or derivatives like 42 which use rearrangements like those showed in in Fig. 8 mechanistically exposed in the same figure [17,21,22]. The alkaline conditions for reactions described in Fig. 6, 7 and 8, are not applicable to substances reactive to alkalis. Instead, the rearrangements can be achieved in neutral media at room temperature by the using of catalytic quantities of Pd-Cl-bis(benzonitrile) complex [17,23]. Alternatively, Hg-bis(trifluoroacetate) in watered THF followed by demercuration by NaBH4 [17,24] or Li-bis(trifluoroacetate) [17,25] can be used.

Comments
In Fig. 8, mechanism A follows a strict pericyclic cyclisation concept which includes the overlapping of orbitals p contiguous, thus the forming and extinction of covalent bonds (π and o) are simultaneous. Under such optic, a transition state in a chair-like shape is formed and this transition state conducts to compound 42'. A 3D model of 42' has been constructed using Prentice Hall Framework Molecular Models (photo in Fig. 8). This 3D model (42') exhibits a strong steric hindrance between the carbonyl π system and the C9=C10 bond (numbering adopted from decaline structure). Thus if a pericyclic process takes place, the "endo" carbonyl conformer results (see Fig 8. Mechanism A). Since this "endo" form implies high instability due to the hindrance of carbonyl and C9=C10, then the system has to evolve somehow towards a more stable conformer, namely the conformer "exo" or compound 42. Mechanism B of Fig. 8 shows how the ionic process can easily conduct to the conformer "exo" when after formed the carbanion and the carbocation a simple rotation and ulterior overlapping of sp2 -> sp3 orbitals forms a new covalence (Csp27, Csp28 -> Csp37-Csp38). We have two options, firstly, a pericyclic (or more precisely, sigmatropic) mechanism that conducts to the strained and hence instable product 42', which should be forced somehow to evolve to the more stable conformer "exo", but without cycle excision (Fig. 8, A.), and alternatively, the ionic process which seems more feasible and that easily affords the more relaxed and stable product 42 (Fig. 8, B.). The couple of π systems composed by the carbonyl group and the C3=C4 double bond, present coordination (parallelism of the four p orbitals) in compound 42 ("exo" form in Fig. 8). The other double bond C9=C10 presents as well parallelism of orbitals p with the other two π systems in 42 ("exo") even though they are not coordinated indeed. Compound 42' ("endo" in Fig. 8) presents the C=O and C3=C4 in coordination (parallelism of the four p orbitals), however from the model, the hindrance between the carbonyl and C9=C10 is so evident that it induces us thinking that such conformer is highly improbable; the parallelism of the three tt systems is also present in 42'. These aspects of the reaction demand a longer meditation and analysis about the pericyclic concept of the Claisen rearrangement versus the ionic alternative.
The Claisen rearrangement also takes place with sulfur compounds. Thus, allyl vinyl sulfides can convert by hydrolysis into y<5-unsaturated carbonyl compounds [17,26]. A remarkable feature of this reaction regards an alkylation that can be done over the carbon contiguous to the Sulfur just before the rearrangement occurs, as for instance with the synthesis of 4-tridecenal (43) from allyl vinyl sulfide (Fig. 9) [26].

Comments
Again, as well as for the precedent examples we face the dichotomy of two mechanisms, this time less mutually contradictory due to the equal facility to afford the final result (Fig. 9). Now, there aren't implied any neither stereochemical nor conformational isometry.
Another reaction similar to the rearrangement of allyl vinyl sulfides is the synthesis of allylsulphonium ylids by means of the [2,3]sigmatropic rearrangement. This is the case for the formation of y-cyclocitral, 44, [26]. This reaction and its mechanistic interpretation is shown in Fig. 10.

Comments
This general mechanism (Fig. 10) consists of the following steps: the allyl vinyl sulfide suffers an excision of a good leaving group at the α position of sulfur. The carbanion formed thus is less basic than sulfur and this last attacks as nucleophile over the carbon directly linked to an alkyl halide to establish a new S-C bond. At this stage we are in the presence of a bipolar species which collapses into an allylsulphonium ylid by means of a [2,3] sigmatropic rearrangement which can be envisaged as a pericyclic or an ionic process.
The synthesis of 44 implies the nucleophilic attack of sulfur from dithiane (C4H8S2) to the alkyl bromide to expel this last and establish a new S-C bond with the consequent sulfur cation formation. The treatment of the substrate with the strong base n-butane lithium, extracts an acidic hydrogen of the methylene vicinal to both sulfur atoms of the dithiane cation. The carbanion obtained converts the intermediate in a bipolar species. This last is in equilibrium with the other form of this intermediate, namely the dithiane unsaturated. Two consecutive 120° rotations of the CH-dithiane group with respect to the cyclohexene, places the S=C double bond of the dithiane unsaturated faced to the C=C double bond of cyclohexene. This is a propitious conformation to effect a sigmatropic Claisen rearrangement to afford the adduct: cyclohexene-dithiane. The alternative ionic rearrangement implies delocalization of the π electrons of the double bond S=C to regenerate the dipole which disappears by the nucleophilic attack of the carbanion to one extreme of the C=C of cyclohexene to form a o bond. The end of the process comes with hydrolysis of the dithiane group and the generation of 44.
ACKNOWLEDGEMENT
The authors express their gratitude to Prof. Eduardo Palenque, Department of Physics, Universidad Mayor de San Andrés, for his bibliographic support.
REFERENCES
1. Bravo, J. 2005, The organic chemistry notebook series, a didactical approach. Theoretical mechanistic approach to diasteroselective synthesis of cis-l,2-dialkenylcyclopropanols and subsequent oxy-Cope rearrangement by Jin Kun Cha et al, Rev.Bol. Quim.,23, 1-10. [ Links ]
2. Bravo, J.A., Mollinedo, P., Penarrieta, J.M., Vila, J.L. 2013, Mechanistic views of intramolecular hydroxycyclopropanation of w-vinyl carboxylic esters, Rev. Bol. Quim., 30 (1), 24-41. [ Links ]
3. Bravo, J.A., Vila, J.L. 2014, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part I; the organic chemistry notebook series, a didactical approach, n° 3. Rev. Bol. Quim., 31 (1), 61-67. [ Links ]
4. Bravo, J.A., Vila, J.L. 2015, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part II; the organic chemistry notebook series, a didactical approach, n° 4, Rev. Bol. Quim., 32 (1), 15-23. [ Links ]
5. Vila, J.L., Bravo, J.A. 2015, Synthesis of alkenes by fragmentation reactions; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 5, Rev. Bol. Quim., 32 (2), 37-44. [ Links ]
6. Bravo, J.A, Vila, J.L. 2015, Synthesis of alkenes by oxidative decarboxylation of carboxylic acids; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 6, Rev. Bol. Quim., 32 (3), 45-52. [ Links ]
7. Bravo, J.A., Vila, J.L. 2015, Synthesis of alkenes from ketones via arylsulphonyl-hydrazones; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 7, Rev. Bol. Quim., 32 (4), 82-89. [ Links ]
8. Bravo, J.A., Vila, J.L. 2015, Stereospecific synthesis of alkenes from 1,2-diols; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 8, Rev. Bol. Quim., 32 (5), 121-125. [ Links ]
9. Bravo, J.A., Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part I; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 9, Rev. Bol. Quim., 33 (1), 27-33. [ Links ]
10. Bravo, J.A, Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part II; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 10, Rev. Bol. Quim., 33 (2), 95-103. [ Links ]
11. Bravo, J.A, Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part III; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 11, Rev. Bol. Quim., 33 (3), 127-133. [ Links ]
12. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 174. [ Links ]
13. Rhoads, S.J., Raulins, N.R., The Claisen and the Cope Rearrangement, In: Organic Reactions, vol. 22, ed. by Dauben, W.G. 1975, Wiley, New York, 22, 1-254. [ Links ]
14. Wehrli, R., Bellus, D., Hansen, H.J., Schmidt, H. 1976, Cope rearrangement - reaction with a manifold mechanism, Chimia, 30 (9), 416-423. [ Links ]
15. Overman, L.E., Renaldo, A.F. 1983, Palladium Dichloride Catalyzed Cope Rearrangements of Functionalized Acyclic 1,5-Dienes, Tetrahedron Letters, 24, 3757-3760. [ Links ]
16. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 175. [ Links ]
17. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 176. [ Links ]
18. Evans, D.A., Golob, A.M. 1975, [3,3] Sigmatropic rearrangements of 1,5-diene alkoxides, J. Am. Chem. Soc, 97, 4765-4766. [ Links ]
19. Evans, D.A., Nelson, J.V. 1980, A stereochemical study of the [3,3]-sigmatropic rearrangement of l,5-diene-3-alkoxides. Applications to the streoselective synthesis of (±)-juvabione, J. Am. Chem. Soc, 102, 774-782. [ Links ]
20. Belles, X. 1987, Control de plagas de insectos con agentes hormonales endocrinos, Folia Entomológica Mexicana, 72, 121-160. [ Links ]
21. Still, W.C. 1977, An expeditious routeto the germacranes. Total synthesis of (.+-.)-acoragermacrone and (.+-.)-preisocalamendiol J. Am. Chem. Soc, 99, 4186-4187. [ Links ]
22. Still, W.C. 1979, (.+-.)-Periplanone-B. Total synthesis and structure of the sex excitant pheromone of the American cockroach J. Am. Chem. Soc, 101, 2493-2495. [ Links ]
23. Bluthe, N., Malacria, M., Gore, J. 1983, Transposition d'oxy-cope a temperature ambiante catlysee par le dichlorure de palladium-bisbenzonitrile, Tetrahedron Lett., 24, 1157-1160. [ Links ]
24. Bluthe, N., Malacria, M., Gore, J. 1982, Reactions des hexadiene-1,5 ols-3 avec le trifluoroacetate mercurique; transposition de oxy-cope a temperature ambiante, Tetrahedron Lett., 23, 4263-4266. [ Links ]
25. Bluthe, N., Malacria, M., Gore, J. 1984, Transposition oxy-cope assistee par le trifluoroacetate mercurique en quantite stoechiometrique et en quantite catalytique, Tetrahedron Lett., 40, 3277-3284. [ Links ]
26. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 177. [ Links ]